Title
题目
Cooperative multi-task learning and interpretable image biomarkers for glioma grading and molecular subtyping
用于胶质瘤分级和分子分型的协作多任务学习与可解释影像生物标志物
01
文献速递介绍
胶质瘤是最常见的原发性脑肿瘤,其治疗方案制定和预后评估高度依赖于肿瘤的组织学分级及分子分型,特别是异柠檬酸脱氢酶(IDH)突变和 1p/19q 共缺失状态(Wesseling & Capper, 2018)。目前,组织活检仍然是确定胶质瘤分级和类型的主要方法。然而,活检是一种侵入性操作,可能带来一定的外科风险,如出血、感染和神经功能损伤等。此外,由于胶质瘤的高度异质性,单次活检可能无法全面反映整个肿瘤的特征,可能导致不完整或不准确的诊断(Han et al., 2020)。因此,探索非侵入性的胶质瘤组织学分级和分子分型方法已成为研究热点。
2. 胶质瘤的非侵入性诊断方法
目前,结合多参数磁共振成像(MRI)与机器学习技术的非侵入性胶质瘤诊断方法已被广泛研究。在早期,影像组学(radiomics)是最具代表性的非侵入性方法之一(Lambin et al., 2017; Yu et al., 2017; Tian et al., 2018; Bae et al., 2018)。该方法通过从肿瘤区域提取大量人工设计的特征,并利用机器学习模型进行胶质瘤分级或分型。然而,这类方法的预测准确性受限于人工特征的有效性。此外,影像组学方法通常需要精确勾画感兴趣区域(ROI),其性能高度依赖于分割结果。随着深度学习方法的快速发展,利用深度学习模型直接从原始多参数 MRI 图像中预测胶质瘤分级和分型已成为一种替代方案。最初的尝试是直接使用针对自然图像分类设计的基础网络进行胶质瘤分类(He et al., 2016; Huang et al., 2017; Howard et al., 2017; Zhang et al., 2018; Gao et al., 2021; Ding et al., 2021)。例如,Nalawade 等(2019)利用 DenseNet 从未经分割的 T2 加权图像中预测 IDH 突变状态,证明了深度特征在提高胶质瘤分类性能方面的优势。为了克服样本规模对预测性能的影响,Jiang 等(2022)基于预训练的 SE-ResNeXt 模型提出了多种迁移学习策略,以提高分类准确性和训练效率。然而,由于 SE-ResNeXt 模型的庞大参数量,该方法计算成本仍然较高。为了解决计算成本问题,Dutta 等(2024)提出了一种基于轻量级全局注意力模块的多尺度卷积网络用于多类别胶质瘤预测,该方法在减少参数量的同时保持较高的预测准确性。但该方法在外部验证集上的泛化能力尚未充分评估。尽管修改卷积神经网络(CNN)架构或训练策略可以改善预测效果,但这些方法忽略了多模态数据中固有的互补信息,这可能限制其预测能力。因此,近年来提出了多模态胶质瘤分类方法,通过有效融合多模态特征,显著提升预测性能。例如,Matsui 等(2020)使用 ResNet 提取 T1 加权、T2 加权和 PET 图像的特征,并简单地将这些特征与临床信息结合,以预测胶质瘤的分子亚型。然而,由于未设计专门的特征融合策略,该方法存在较严重的过拟合问题。为了解决该问题,一些基于注意力机制的多模态分类模型相继提出(Xu et al., 2024; Cluceru et al., 2022)。典型方法包括 AGGN(Wu et al., 2023)和基于 Swin-transformer 的方法(Pacal, 2024)。前者在空间金字塔模型中引入注意力机制,以融合不同序列的多尺度互补特征,提高了胶质瘤分级性能;后者采用混合滑动窗口多头自注意力模块以有效融合多模态信息,并利用残差 MLP 层替换 Transformer 块中的 MLP 层以提升预测精度。然而,尽管这些注意力机制方法在单中心数据集上表现良好,在多中心数据集上仍可能受异质背景信息的影响,导致泛化能力受限。
为提高泛化能力,一种改进策略是将肿瘤区域的先验知识引入分类网络,例如利用肿瘤边界框(He et al., 2021; Yan et al., 2022; Tandel et al., 2022)或肿瘤掩码(Ning et al., 2021; Wei et al., 2023)限制模型输入仅包含肿瘤区域,以抑制异质背景信息的影响。然而,这些方法需要事先标注肿瘤掩码,实际应用中并不总是可行。为克服这一问题,提出了两阶段方法(Naser & Deen, 2020; Tripathi & Bag, 2022; Fang & Jiang, 2024),即先检测或分割肿瘤区域,再进行分类预测。尽管此类方法在一定程度上提高了模型的泛化能力,但计算量大、耗时较长。此外,第一阶段分割或检测的错误可能会显著影响第二阶段的分类准确性。因此,胶质瘤分割与分类的多任务学习框架逐渐受到关注。例如,Cheng 等(2022)提出了 MTTU-Net 框架,该框架结合 CNN 和 Transformer 结构,同时预测 IDH 突变状态并分割肿瘤区域。然而,由于密集语义预测(分割)与全局语义预测(分类)之间的信息鸿沟,该方法的 IDH 预测准确性未能达到理想水平。不同于 MTTU-Net,MFEFNet(Zhang et al., 2024)采用 U-Net 分割胶质瘤,同时利用一个精心设计的编码器提取可区分特征进行 IDH 预测。该方法通过双注意力机制融合 U-Net 编码特征与分类编码特征,提高了 IDH 预测的准确性。然而,该方法仍然专注于单一分子分型任务,忽略了胶质瘤不同分子分型任务与分级任务之间的相互作用,可能限制其在临床应用中的有效性(Molinaro et al., 2019; Zhang et al., 2021b)。目前,能够同时执行多个胶质瘤分类任务与分割任务的方法仍较少。Decuyper 等(2021)和 van der Voort 等(2022)提出了多任务协作模型,同时执行胶质瘤分割、分级及 IDH 和 1p/19q 状态预测。然而,由于密集语义预测任务(分割)与全局语义预测任务(分类)之间的信息差异,难以保证分割与分类的同时准确性。此外,这些方法仅挖掘了多任务之间的共享特征,未充分探索任务特异性特征,限制了泛化能力,同时缺乏可解释性,无法提供与模型决策相关的影像生物标志物。
3. 研究贡献
为解决上述挑战,我们提出了一种新型的协作多任务学习网络(CMTLNet),并探索用于胶质瘤诊断的可解释影像生物标志物。主要贡献如下:
无分割的肿瘤特征感知(SFTFP)模块,通过自定义的肿瘤相关/非相关掩码-图像对,以二分类方式提取隐式肿瘤特征,避免了分割与分类任务之间的协同问题。任务共享特征提取,通过大余弦间隔损失约束类内特征一致性和类间特征区分性,以优化多任务共享特征的学习。
基于正交投影和条件分类的任务特异性特征提取模块,进一步增强特异性信息提取能力。独有-共享特征协同分类模块,通过空间与通道注意力融合特征,提升多任务预测性能。结合 SHAP 值和相关性分析,探索深度特征引导的可解释影像组学生物标志物,提高模型可解释性和临床实用性。
Aastract
摘要
Deep learning methods have been widely used for various glioma predictions. However, they are usually taskspecific, segmentation-dependent and lack of interpretable biomarkers. How to accurately predict the gliomahistological grade and molecular subtypes at the same time and provide reliable imaging biomarkers is stillchallenging. To achieve this, we propose a novel cooperative multi-task learning network (CMTLNet) whichconsists of a task-common feature extraction (CFE) module, a task-specific unique feature extraction (UFE)module and a unique-common feature collaborative classification (UCFC) module. In CFE, a segmentation-freetumor feature perception (SFTFP) module is first designed to extract the tumor-aware features in a classificationmanner rather than a segmentation manner. Following that, based on the multi-scale tumor-aware featuresextracted by SFTFP module, CFE uses convolutional layers to further refine these features, from which the taskcommon features are learned. In UFE, based on orthogonal projection and conditional classification strategies,the task-specific unique features are extracted. In UCFC, the unique and common features are fused withan attention mechanism to make them adaptive to different glioma prediction tasks. Finally, deep featuresguided interpretable radiomic biomarkers for each glioma prediction task are explored by combining SHAPvalues and correlation analysis. Through the comparisons with recent reported methods on a large multi-centerdataset comprising over 1800 cases, we demonstrated the superiority of the proposed CMTLNet, with the meanMatthews correlation coefficient in validation and test sets improved by (4.1%, 10.7%), (3.6%, 23.4%), and(2.7%, 22.7%) respectively for glioma grading, 1p/19q and IDH status prediction tasks. In addition, we foundthat some radiomic features are highly related to uninterpretable deep features and that their variation trendsare consistent in multi-center datasets, which can be taken as reliable imaging biomarkers for glioma diagnosis.The proposed CMTLNet provides an interpretable tool for glioma multi-task prediction, which is beneficial forglioma precise diagnosis and personalized treatment.
深度学习方法已广泛应用于各种胶质瘤预测任务。然而,这些方法通常具有任务专一性、依赖分割且缺乏可解释的影像生物标志物。如何在准确预测胶质瘤组织学分级和分子亚型的同时,提供可靠的影像生物标志物仍然是一个挑战。为此,我们提出了一种新型的协作多任务学习网络(CMTLNet),该网络由任务共享特征提取(CFE)模块、任务特异性独有特征提取(UFE)模块和独有-共享特征协同分类(UCFC)模块组成。
在 CFE 模块中,我们首先设计了一个无分割的肿瘤特征感知(SFTFP)模块,以分类方式而非分割方式提取肿瘤感知特征。随后,基于 SFTFP 模块提取的多尺度肿瘤感知特征,CFE 通过卷积层进一步优化这些特征,从中学习任务共享特征。在 UFE 模块中,我们基于正交投影和条件分类策略提取任务特异性的独有特征。在 UCFC 模块中,我们利用注意力机制融合独有和共享特征,使其适应不同的胶质瘤预测任务。
最后,我们结合 SHAP(SHapley Additive exPlanations)值和相关性分析,探索由深度特征引导的可解释影像组学生物标志物,以支持各个胶质瘤预测任务。在包含 1800 余例的多中心数据集上,我们将 CMTLNet 与近期报道的方法进行了比较,结果表明 CMTLNet 具有显著优势,其在验证集和测试集上的 Matthews 相关系数(MCC)平均提高了 4.1% 和 10.7%(胶质瘤分级),3.6% 和 23.4%(1p/19q 状态预测),2.7% 和 22.7%(IDH 状态预测)。此外,我们发现部分影像组学特征与深度特征高度相关,且其变化趋势在多中心数据集中保持一致,这些特征可作为胶质瘤诊断的可靠影像生物标志物。所提出的 CMTLNet 为胶质瘤多任务预测提供了一种可解释的工具,有助于精准诊断和个性化治疗。
Conclusion
结论
We proposed a cooperative multi-task learning framework (CMTLNet), which not only can predict simultaneously glioma grade andmolecular subtypes, but also can provide interpretable imaging biomarkers for each task. It first uses a SFTFP module to extract tumorrelated features from unsegmented images, overcoming the issue ofsegmentation-dependence in the inference phase. Then, it employs aCFE module to extract across-task-common features, and a UFE moduleto extract task-specific features based on orthogonal projection andconditional guidance strategies. Finally, a UCFC module is used toadaptively fuse unique and common features to realize multiple taskprediction. In addition, to obtain explainable imaging biomarkers foreach task, we combined SHAP, correlation analysis methods and radiomic features to explore the biomarkers. The results showed that theproposed CMTLNet significantly improves the prediction performancein multicenter glioma grading and molecular subtyping. The firstorder features of T1C images, as well as first-order features of T1Cimages after exponential, logarithmic, and wavelet transformations canbe taken as the biomarkers for glioma grading and subtyping. Theproposed CMTLNet can be taken as an interpretable tool for gliomamulti-task prediction, providing an useful auxiliary tool for gliomaprecise diagnosis and personalized treatment.
我们提出了一种协作多任务学习框架(CMTLNet),该框架不仅能够同时预测胶质瘤分级和分子分型,还能够为每个任务提供可解释的影像生物标志物。
首先,CMTLNet 采用SFTFP 模块从未经分割的图像中提取肿瘤相关特征,从而克服了推理阶段对分割的依赖性。接着,CFE 模块提取跨任务共享特征,而UFE 模块则基于正交投影和条件引导策略提取任务特异性特征。最后,UCFC 模块自适应地融合独有特征和共享特征,以实现多任务预测。
此外,为了获得可解释的影像生物标志物,我们结合了 SHAP 分析、相关性分析 和 影像组学特征提取 方法,探索各任务的生物标志物。实验结果表明,所提出的 CMTLNet 在多中心胶质瘤分级和分子分型任务中显著提高了预测性能。研究还发现,T1C 图像的一级特征,以及 T1C 图像经过指数变换、对数变换和小波变换后的一级特征,可作为胶质瘤分级和分型的影像生物标志物。总体而言,所提出的 CMTLNet 可作为可解释的胶质瘤多任务预测工具,为胶质瘤精准诊断和个性化治疗提供有力的辅助支持。
Results
结果
4.1. Comparison with existing glioma prediction methods Table 2 gives the mean ± std of several evaluation metrics obtainedby different methods on five-fold cross validation datasets for eachglioma prediction task. We can observe that the mean metrics of proposed method CMTLNet on all prediction tasks are significantly higherthan the other comparison methods (𝑝 ≤ 0.05 and the corresponding95% confidence intervals does not overlap with the others except forthe AUC of LHG classification task (Table A1 of the Appendix 2).Even though some models can obtain comparable performance as ourmethod in terms of a given metric, they cannot balance the othermetrics. For instance, in LHG prediction task, the mean AUC of ARMNetis a little bit higher than our CMTLNet (not significantly, 𝑝 ≥ 0.05), butits ACC, F1-score, and MCC are significantly (𝑝 ≤ 0.001) reduced by2.8%, 2.5% and 6.0% respectively compared with our method. In contrast, the proposed CMTLNet can improve these metrics simultaneously;compared with the existing methods, its average ACC, F1-score, MCC,and AUC on three predication tasks are improved by at least 1.7%,2.4%, 4.9% and 1.4% respectively.To show the generalization ability of the proposed model, we givein Table 3 the evaluation metrics of different methods on the externaltest sets from different centers. It can be found that, except on theREMBRANDT data set, the AUC of the proposed CMTLNet for LHGprediction task is slightly lower than that of the model of Decuyperet al. (0.59%, 𝑝 ≤ 0.001). On the UCSF-PDGM data set, its AUC for1p/19q prediction task is lower than existing model (5.6% with 𝑝 ≥0.05 lower than the highest AUC). The proposed CMTLNet achievesthe best evaluation metrics for all prediction tasks on all external testsets (Table 3 (Mean, the last panel)). Specifically, compared to thecorresponding sub-optimal model, the average MCC of CMTLNet onmulti-center test sets is significantly (𝑝 ≤ 0.001) increased by 10.7%,23.4% and 22.7% respectively for LHG, 1p/19q and IDH predictiontasks, the corresponding improvement of ACC reaches 7.0%, 0.9% and10.8% respectively with 𝑝 ≤ 0.001, and the average F1-score for thesethree tasks is also significantly (𝑝 ≤ 0.001) improved by 6.4%, 23.6%and 10.6%. Such statistical differences can also be found in Table A2of Appendix 2, besides AUC, the confidence intervals of all the othermetrics obtained by our method do not overlap with the intervalsderived from the rest methods.Since our work is to predict the status of LGG/HGG, 1p/19q codeletion, and IDH mutations in gliomas simultaneously, we have alsoprovided the task-averaged ROC curves on both validation and test setsin Fig. 3 to comprehensively assess the multi-task prediction performance of different models. We can see that, on the validation sets, ourmethod (black line) outperforms slightly the comparison methods interms of average ROC and AUC. However, on the external test sets,our average ROC curve is much better than the others, illustratingthe superiority of the proposed CMTLNet in generalized multi-taskprediction.
4.1 与现有胶质瘤预测方法的比较
表 2 显示了在 五折交叉验证数据集 上,不同方法在各个胶质瘤预测任务中的评估指标(均值 ± 标准差)。可以观察到,在所有预测任务中,所提出的 CMTLNet 方法的平均评估指标显著高于其他对比方法(p ≤ 0.05),且其 95% 置信区间(CI)与其他方法不重叠(除 LHG 分类任务的 AUC,具体见 附录 2 表 A1)。尽管某些模型在某一特定指标上可获得与 CMTLNet 相当的性能,但它们无法在所有评估指标上同时保持平衡。例如:在 LHG 预测任务中,ARMNet 的平均 AUC 略高于 CMTLNet(但无统计学差异,p ≥ 0.05),然而,其 ACC(准确率)、F1-score 和 MCC(Matthews 相关系数)分别比 CMTLNet 低 2.8%、2.5% 和 6.0%(p ≤ 0.001),表明其整体预测能力受限。相比之下,CMTLNet 可同时提升所有关键指标,在三个预测任务中,其 ACC、F1-score、MCC 和 AUC 的平均提升幅度至少分别达到 1.7%、2.4%、4.9% 和 1.4%。为了评估所提出模型的泛化能力,表 3 展示了不同方法在多中心外部测试集上的评估指标。从结果来看:除了 REMBRANDT 数据集外,CMTLNet 在 LHG 预测任务上的 AUC 略低于 Decuyper 等人提出的方法(下降 0.59%,p ≤ 0.001)。在 UCSF-PDGM 数据集上,CMTLNet 在 1p/19q 预测任务上的 AUC 低于现有最优模型(下降 5.6%,p ≥ 0.05)。
总体而言,CMTLNet 在所有外部测试集上的所有预测任务中均获得了最佳评估指标(见表 3 末列Mean)。MCC 提升幅度(p ≤ 0.001):LHG 10.7%,1p/19q 23.4%,IDH 22.7%ACC 提升幅度(p ≤ 0.001):LHG 7.0%,1p/19q 0.9%,IDH 10.8%F1-score 提升幅度(p ≤ 0.001):LHG 6.4%,1p/19q 23.6%,IDH 10.6%此外,从 附录 2 表 A2 的统计分析结果来看,除了 AUC 以外,CMTLNet 取得的所有其他评估指标的置信区间均与其他方法无重叠,进一步表明 CMTLNet 具有较强的统计学优势。多任务预测性能评估,由于本研究的目标是同时预测 LGG/HGG(低级别胶质瘤/高级别胶质瘤)、1p/19q 共缺失状态和 IDH 突变状态,因此在图 3 中,我们绘制了不同模型在验证集和测试集上的任务平均 ROC 曲线,以全面评估多任务预测的整体性能:在验证集上,CMTLNet(黑色曲线)在平均 ROC 和 AUC 方面略优于其他方法。在外部测试集中,CMTLNet 的 平均 ROC 曲线明显优于其他方法,这进一步表明 CMTLNet 在泛化性和多任务预测能力方面的优势。综上,CMTLNet 不仅能在多任务预测中提升整体性能,还能在外部测试数据上展现良好的泛化能力,为胶质瘤的精准诊断和个性化治疗提供了有力的技术支持。
Figure
图

Fig. 1. Illustration of data repartition and train-validation sets splitting for each task. (a) Repartition the samples in MI-20, EGD and LGG-1p/19q datasets into several subsetsbased on their task labels. (b) Illustrate the train and validation sets splitting manner for each task、
图 1. 数据重划分及各任务的训练集-验证集划分示意图。 (a) 根据任务标签,将 MI-20、EGD 和 LGG-1p/19q 数据集中的样本划分为多个子集。 (b) 展示每个任务的训练集与验证集的划分方式。
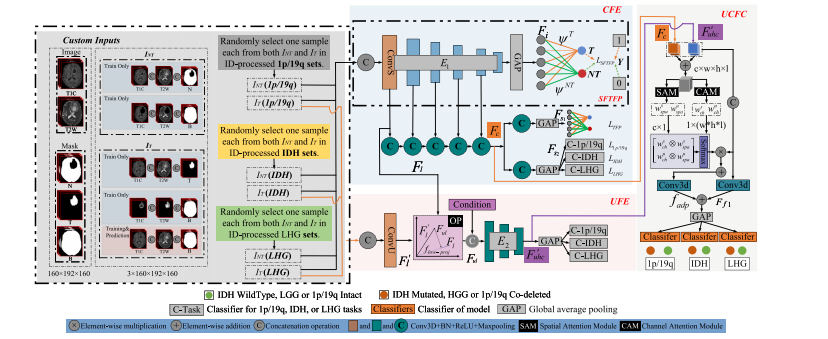
Fig. 2. The overall workflow of the proposed CMTLNet. It consists of three main parts: The first part is task-common feature extraction (CFE) module which is used to extractthe shared features of three glioma prediction tasks; the second part is task-unique feature extraction (UFE) module which is responsible for extracting the task-specific features;the last part is unique-common feature collaborative classification (UCFC) module which fuses the task-common and task-specific features with spatial and channel attentions andthen predicts the multiple glioma classification tasks.
图 2. 所提出的 CMTLNet 的整体工作流程。 该网络由三大核心模块组成:任务共享特征提取(CFE)模块:用于提取三种胶质瘤预测任务的共享特征;
任务特异性特征提取(UFE)模块:负责提取各任务独有的特征;独有-共享特征协同分类(UCFC)模块:通过空间和通道注意力机制融合任务共享和任务特异性特征,并最终完成多任务胶质瘤分类预测。

Fig. 3. Average ROC curves across all tasks for the validation set (a) and test set (b)
图 3. 所有任务的平均 ROC 曲线: (a) 验证集的平均 ROC 曲线;(b) 测试集的平均 ROC 曲线。

Fig. 4. Comparison of the predictive performance of different ablation models in different tasks. The ‘mean’ represents the average performance across multiple tasks.
图 4. 不同消融模型在不同任务中的预测性能比较。“mean” 代表多个任务的平均性能。
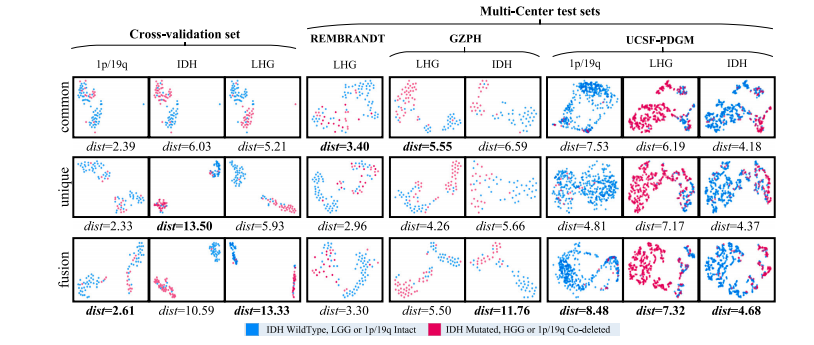
Fig. 5. T-SNE visualization of the unique, common and fused features for each task across different datasets. ‘dist’ denotes the ratio of cosine distance of inter-class samples tothat of intra-class samples.
图 5. 不同数据集中,各任务的独有特征、共享特征及融合特征的 T-SNE 可视化。“dist” 表示类别间样本的余弦距离与类别内样本的余弦距离的比值。
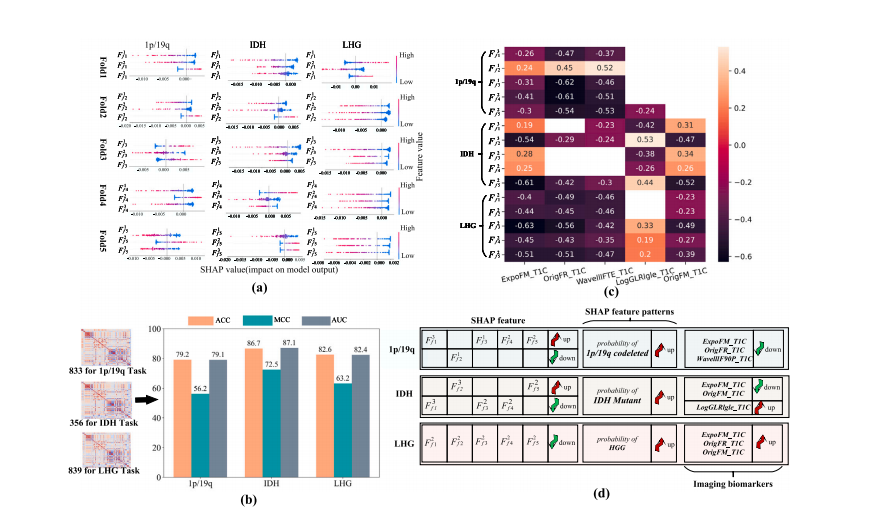
Fig. 6. The relevant results for interpretable imaging biomarkers mining. (a) SHAP summary plot for the top 3 deep features of each task on five-fold validation sets. The samplewith higher feature SHAP value tends to be predicted as the positive class (HGG, IDH mutations, and 1p/19q co-deletions in three tasks). (b) Selected possible radiomic featuresrelated to each task and the classification performance of these features. © Correlation heatmap between deep features selected with SHAP and the potential image biomarkersfor each glioma prediction task. Blank cell indicates no significant correlation between two features (𝑃 > 0.05). (d) The variation trends of deep features and image biomarkers fordifferent task predictions are summarized. The full name of biomarkers can be found in the Appendix 1.
图 6. 可解释影像生物标志物挖掘的相关结果。 (a) SHAP 综述图:展示五折交叉验证集中每个任务排名前 3 的深度特征。SHAP 值较高的样本更可能被预测为阳性类别(HGG、IDH 突变和 1p/19q 共缺失)。 (b) 选定的潜在影像组学特征:与每个任务相关,并展示其分类性能。 © 相关性热图:显示 SHAP 选出的深度特征与各胶质瘤预测任务的潜在影像生物标志物之间的相关性。空白单元格表示两者之间无显著相关性(P > 0.05)。 (d) 深度特征与影像生物标志物的变化趋势:总结不同任务预测中深度特征与影像生物标志物的变化模式。完整的生物标志物名称可见附录 1。

Fig. 7. Scatterplot of imaging biomarkers for real positive (red points) and negative samples (blue points) in the validation (a) and multi-center test sets (b) of each task.
图 7. 影像生物标志物的散点图,区分真实阳性样本(红色点)和真实阴性样本(蓝色点)。 (a) 验证集中各任务的散点分布; (b) 多中心测试集中各任务的散点分布。
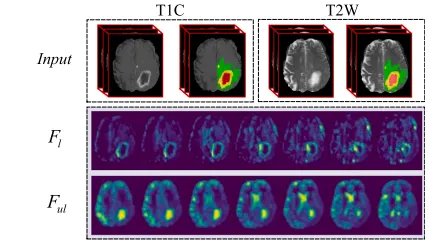
Fig. 8. Visualization of across-task-common feature (𝐹𝑙 ) and the unique featureextracted with orthogonal projection disentanglement (𝐹𝑢𝑙) for a randomly selectedsample. The green area marked in the input images represents the tumor edema region;the yellow and red area signifies the tumor core and the necrotic region, respectively
图 8. 跨任务共享特征 (𝐹𝑙 ) 与通过正交投影解耦提取的独有特征 (𝐹𝑢𝑙 ) 的可视化展示,示例为随机选取的样本。 输入图像中标记的绿色区域代表肿瘤水肿区,黄色和红色区域分别表示肿瘤核心区和坏死区。
Table
表
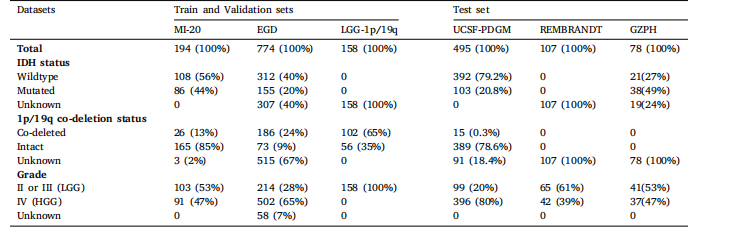
Table 1Clinical characteristics of the samples in train, validation and test sets.
表 1 训练集、验证集和测试集中样本的临床特征。

Table 2The average quantitative evaluation metrics (mean ± standard deviation) of different prediction models on five-fold cross-validation sets.
表 2 不同预测模型在五折交叉验证集上的平均定量评估指标(均值 ± 标准差)。
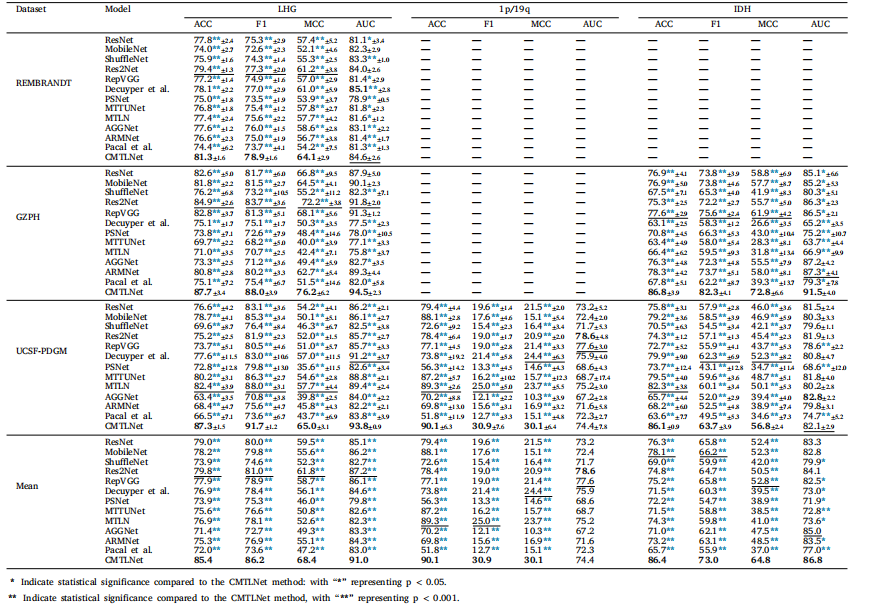
Table 3The average quantitative evaluation metrics (mean ± standard deviation) for different prediction models on an external test sets
表 3 不同预测模型在外部测试集上的平均定量评估指标(均值 ± 标准差)。